


Genomics is a transformative and data-rich field that has experienced tremendous growth over the past 12 years. With the advent of Next-Generation Sequencing (NGS) platforms in 2005, an exponential amount of data has been generated and deposited into the National Center for Biotechnology Information databases [1]. Genomics provides valuable research opportunities to help describe the function, structure, and evolution of all living organisms. For the Chemical and Biological Defense Program (CBDP), it has provided Department of Defense (DoD) laboratories with a powerful tool that helps characterize biological threat agents and an abundance of information that shapes detection, diagnostics, therapeutics, and prophylaxis capabilities.
In 2003, initial sequencing of the entire human genome cost approximately $2.7 billion [2]. Fifteen years later, what once took billions of dollars to accomplish, now can cost less than $1,000 [3]. Today, valuable data can be generated in a relatively cost-effective manner from numerous sequencing platforms available on the market. A steady and continuous decrease in genome sequencing costs is projected, which may lead to: 1) an overwhelming abundance of data and 2) increased opportunities to utilize NGS to address various research topics in a cost-effective manner [4]. The decrease in genome sequencing costs and the proliferation of various next generation sequencing technologies show great promise in improving force health protection.
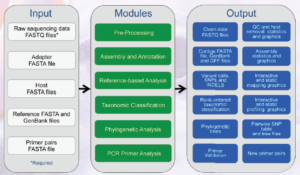
Figure 1. An overview of the EDGE Bioinformatics workflow and modules [7]
In addition to the sheer amount of data being generated, this NGS data is often not readily actionable or informative as it requires significant analyses and data processing. In order to address this big data challenge, and ultimately best support the warfighter, we must overcome a significant technology gap—how can the U.S. military rapidly analyze all the resulting sequencing data without a team of bioinformaticians?
Empowering the Development of Genomic Expertise (EDGE) Bioinformatics was developed to address the genomic analysis challenge to effectively utilize NGS for the CBDP mission. For example, the ability to quickly analyze genetic data may allow the U.S. military to determine if a warfighter has been exposed to a biological warfare agent; determine whether the causative agent is a virus or bacteria; inform medical countermeasures such as prescribing antibiotics; and provide information on circulating pathogens and infectious diseases in field-forward locations prior to deployment.
Challenges Using NGS
In 2013, DoD began equipping laboratories located in the U.S. and abroad with cutting-edge equipment, such as the Illumina MiSeq next-generation sequencer and server systems, in an effort to improve warfighter protection from biological warfare agents and public health threats. However, the adoption and use of these state-of-the-art NGS technologies have been limited due to numerous complexities involving, but not limited to, sample preparation and workflow bottlenecks associated with genomic analysis. This, coupled with the overwhelming amounts of NGS data generated from a sequencing run, can be a daunting challenge for inexperienced users or those that lack significant bioinformatic training. The U.S. military is not immune to the shortage of bioinformaticians, making the rapid adoption of cutting-edge diagnostic equipment even more difficult [6].
Solving the Big Data Analysis Challenge
To readily address the analysis bottleneck issues, the Joint Science and Technology Office (JSTO) at the Defense Threat Reduction Agency (DTRA) has collaborated with Los Alamos National Laboratory (LANL) and the Biological Defense Research Directorate at the Naval Medical Research Center to develop and pilot the EDGE Bioinformatics platform. This suite of bioinformatic tools was initially tailored to support the Illumina MiSeq, providing a variety of modular tools that can analyze genomic data generated by various NGS instruments. Specifically, EDGE Bioinformatics can analyze any raw data in the FASTA/FASTQ file format.
EDGE Bioinformatics has pre-configured workflows or modules (see Figure 1) for analyzing genomic data, identifying relevant genes of interest, and creating reports and graphics with an easy-to-use web-interface [7]. The latest version of EDGE (Version 1.5) is built around a collection of more than 50 publicly available open-source software packaged into seven modular workflows: pre-processing; assembly and annotation; reference-based analysis; taxonomic classification; phylogenetic analysis; specialty gene analysis; and polymerase chain reaction primer analysis [7]. An operator can select modules individually or run them in any number of combinations to address particular analysis needs. Specific modules used in the EDGE Bioinformatics suite are dependent on the user’s scientific hypotheses and experimental needs. Additional tools and modules can be developed or incorporated to best support the variety of analyses needed for applications across DoD. Many of these applications can be leveraged by other government agencies that have similar scientific questions and require similar bioinformatics solutions.
Bioinformatics can be used to detect virulence factors, determine bacterial strain type, combat the emerging threat of antimicrobial resistance, and identify dangerous pathogens [8]. EDGE Bioinformatics relies on several modules, which have numerous tools within each to readily analyze and make sense of the wealth of data. One popular module within the EDGE Bioinformatics suite is the taxonomic classification module, which currently uses numerous taxonomy tools such as Genomic Origins Through Taxonomic CHAllenge (GOTTCHA) to help identify biological organisms such as pathogens in environmental and/or clinical samples using sequencing data.
GOTTCHA was designed by LANL researchers (developers of the EDGE Bioinformatics suite) to detect and classify microbes present from a mixed metagenomic sample [9]. In addition to GOTTCHA, EDGE Bioinformatics allows the user to utilize and compare other alternative taxonomy tools (e.g., Kraken, BWA, DIAMOND, etc.) (see Figure 2a). The user may gain confidence in the reliability of his/her results because multiple tools yield the same output (although based on different algorithms).
Taxonomy tools such as GOTTCHA can be used to help identify Bacillus anthracis from a stool sample or help detect Francisella tularensis from air [9]. Alternatively, if the user was also interested in identifying antimicrobial resistance genes or virulence factors, the user would simply use the specialty gene analysis module.

Figure 2. EDGE outputs a variety of files, tables, and graphics that can be viewed on screen or downloaded. For instance, (a) heatmap identifying the Zaire ebolavirus as the most probable
causative agent; and (b) Krona plot view of the same data can also be seen.
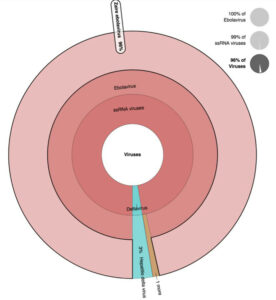
Figure 2. (b) Krona plot view of the same data can also be seen.
According to the 2015 National Action Plan for Combating Antibiotic-resistant Bacteria, antibiotic-resistant bacteria pose an “urgent and serious” threat to both the U.S. and global population [10]. In fact, the DoD was directed to “fund at least one project involving next-generation sequencing technologies or bioinformatics platforms or tools that can be leveraged to improve diagnostics for drug-resistant or multidrug resistant pathogens [10].” EDGE Bioinformatics is addressing this serious threat through the specialty genes analysis module that leverages Short, Better Representative Extract Dataset (ShortBRED) to help identify phylogenetic signatures of antibiotic resistance. ShortBRED is a software tool developed by Harvard University researchers that profiles proteins families of interests from sequencing data [11].
In the specialty gene analysis module, EDGE Bioinformatics can perform either read-based or DNA fragment/contig-based analysis, which the user can easily select to profile protein families associated with antibiotic- resistance or virulence factors (or both) depending on the needs of the user. EDGE Bioinformatics is the first platform designed to allow the user to operate any number of tools (developed by multiple organizations) independently or in parallel using genomic sequencing data.
The only input required from the user is raw sequencing data in the form of a FASTA or FASTQ file, and the EDGE Bioinformatics platform will generate accessible outputs, including text and figures (some featuring user-interactive graphics) [7]. This means no coding experience is required. Instead, a laboratory technician can analyze a sample in as little as minutes to hours (instead of days or weeks) by simply toggling featured options on or off within each module, promptly gaining access to results that can be directly provided to decision-makers. Although EDGE Bioinformatics was designed with non-bioinformaticians in mind, it can easily be customized for use by advanced bioinformaticians with an optional command line interface seen in other platforms such as Galaxy and BaseSpace [12]. These more experienced bioinformaticians can utilize EDGE Bioinformatics to initially run data through the platform and then delve deeper into the NGS data using custom workflows.
EDGE Bioinformatics 2.0
EDGE Bioinformatics Version 2.0 is currently under research and development and is scheduled to be released in 2019. It will include enhanced versions of tools for taxonomy and phylogeny and will also incorporate tools tailored for transcriptomic studies with ribonucleic acid sequencing (RNA seq) as well as software tools designed by the U.S. Army Medical Research Institute of Infectious Diseases (USAMRIID). These USAMRIID-developed tools will assist with pathogen discovery and develop NGSbased laboratory developed tests. Once Version 2.0 becomes available, JSTO will encourage others to incorporate third party workflows and tools. This will provide ample opportunity for collaboration, serving as a way to crowdsource bioinformatic solutions, overcoming the big data analysis challenge of genomics.
Outside of genomics, EDGE Bioinformatics will begin providing solutions to RNA seq/transcriptomic studies that evaluate gene expression. Studying gene expression allows JSTO and DoD to gain a deeper understanding of cellular functions and processes that may contribute to disease. To readily address and enable these types of research studies, EDGE Bioinformatics developers at LANL are actively working on a tool called Pipeline for Reference-based Transcriptomics (known as PiReT) that will allow researchers to discover and evaluate biomarkers or signatures from host–pathogen interactions. The interplay between host and microbe can provide early indicators/signatures that could in turn prove to be revolutionary in how we diagnose and control diseases [13].
Moving forward, EDGE Bioinformatics will empower the user to incorporate his/her own tools and workflows into the suite. Furthermore, JSTO is investigating the incorporation of tools built for other sequencing platforms in order to accommodate upcoming NGS technologies, such as the Oxford Nanopore Technologies MinION. Eventually, a fully functional EDGE Bioinformatics will allow DoD to develop a Food and Drug Administration cleared and approved NGS-based test for clinical use against biological threat agents.
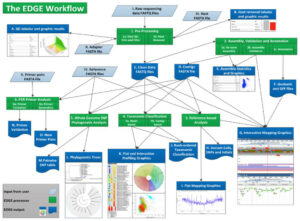
Figure 3. A detailed overview of the EDGE Bioinformatics Version 1 environment
Conclusion
EDGE Bioinformatics was intended for use in DoD laboratories that lack bioinformatics capabilities. However, it is readily being utilized internationally by private industry, health care providers, other government laboratories, and academia. It has been successfully installed in more than 20 DoD and partner-nation laboratories across 11 countries, including a number of Naval Medical Research Units and Centers for Disease Control and Prevention laboratories [12, 14]. DoD is primarily using EDGE Bioinformatics for analyses of microbial genomics and identification of biological threat agents. EDGE Bioinformatics was used to help analyze the sequencing data from the 2014-2016 Ebola Outbreak in West Africa and was more recently used to track diseases such as hantavirus in the Republic of Korea [7, 15]. Ultimately, in order to utilize NGS as a tool to aid medical diagnostics, hardened bioinformatic solutions that readily process and quickly analyze data in a timely fashion will be required. Because of time hindrances, NGS is not yet a premier diagnostic tool but rather a research tool that can provide informative laboratory data that can potentially aid clinicians and influence diagnosis [16]. An accurate medical diagnosis ideally requires a comprehensive amount of information, including patient (i.e., warfighter) history and physical examination and test results.
Future DTRA-JSTO investments regarding host-pathogen interactions will continue to make sequencing a useful research tool that may ultimately aid in the advancement of novel diagnostic capabilities, with the hope that NGS can be used to diagnose ill patients in the future. Until then, EDGE Bioinformatics will continue to be used to help develop diagnostic products and support biological detection capabilities.
References
1. Stephens, Z. D., Lee, S. Y., Faghri, F., Campbell, R. H., Zhai, C., Efron, M. J., . . . Robinson, G. E. (2015). Big data: Astronomical or genomical? PLOS Biology, 13(7). doi:10.1371/journal.pbio.1002195
2. Voelkerding, K. V., Dames, S. A., & Durtschi, J. D. (2009). Next-generation sequencing: From basic research to diagnostics. Clinical Chemistry, 55(4), 641-658. doi:10.1373/clinchem.2008.112789
3. van Dijk, E. L., Auger, H., Jaszczyszyn, Y., & Thermes, C. (2014). Ten years of next-generation sequencing technology. Trends in Genetics, 30(9), 418-426.doi:10.1016/j.tig.2014.07.001
4. Buermans, H. P., & den Dunnen, J. T. (2014). Next generation sequencing technology: Advances and applications. Biochimica et Biophysica Acta (BBA) – Molecular Basis of Disease, 1842(10), 1932–1941. doi:10.1016/j.bbadis.2014.06.015
5. Deurenberg, R. H., Bathoorn, E., Chlebowicz, M. A., Couto, N., Ferdous, M., García-Cobos, S., . . . Rossen, J. W. (2017). Application of next generation sequencing in clinical microbiology and infection prevention. Journal of Biotechnology, 243(10), 16-24. doi:10.1016/j.jbiotec.2016.12.022
6. Chang, J. (2015). Core services: Reward bioinformaticians. Nature, 520(7546),151-152. doi:10.1038/520151a
7. Li, P., Lo, C., Anderson, J. J., Davenport, K. W., Bishop-Lilly, K. A., Xu, Y., . . . Chain, P. S. (2016). Enabling the democratization of the genomics revolution with a fully integrated web-based bioinformatics platform. Nucleic Acids Research, 45(1), 67-80. doi:10.1093/nar/gkw1027
8. Saeb, A. T. (2018). Current bioinformatics resources in combating infectious diseases. Bioinformation, 14(1), 31-35.doi:10.6026/97320630014031
9. Freitas, T. K., Li, P., Scholz, M. B., & Chain, P. S. (2015). Accurate read based metagenome characterization using a hierarchical suite of unique signatures. Nucleic Acids Research, 43(10). doi:10.1093/nar/gkv180
10. The White House. (2015). National Action Plan for Combating Antibiotic-Resistant Bacteria. Retrieved from https://obamawhitehouse.archives.gov/sites/default/files/docs/-national_action_plan_for_combating_antibotic-resistant_bacteria.pdf
11. Kaminski, J., Gibson, M. K., Franzosa,E. A., Segata, N., Dantas, G., & Huttenhower, C. (2015). High-specificity targeted functional profiling in microbial communities with ShortBRED. PLoS Computational Biology, 11(12). doi:10.1371/journal.pcbi.1004557
12. Perkel, J. M. (2017). How bioinformatics tools are brining genetic analysis to the masses. Nature, 543(7643), 137-138.doi:10.1038/543137a
13. Knights, D., Parfrey, L. W., Zaneveld, J., Lozupone, C., & Knight, R. (2011). Human-associated microbial signatures: Examining their predictive value. Cell Host Microbe, 10(4), 292-296. doi: 10.1016/j.chom.2011.09.003
14. Mokashi, V. (2014, March 31). Navy researchers demonstrate bioinformatics capability in Thailand [Web log post]. Retrieved from http://navymedicine.navylive.dodlive.mil/archives/6168
15. Kim, W., No, J. S., Lee, S., Song, D. H., Lee, D., Kim, J., . . . Song, J. (2018). Multiplex PCR−based next-generation sequencing and global diversity of Seoul Virus in humans and rats. Emerging Infectious Diseases, 24(2), 249-257.doi:10.3201/eid2402.171216
16. Lefterova, M. I., Suarez, C. J., Banaei, N., & Pinsky, B. A. (2015). Next-generation sequencing for infectious disease diagnosis and management: A report of the Association for Molecular Pathology. The Journal of Molecular Diagnostics, 17(6), 623-634. doi:10.1016/j.jmoldx.2015.07.004